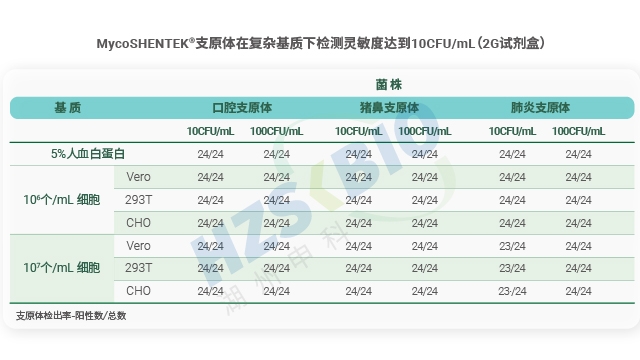
检测限验证是支原体 NAT 方法(核酸扩增法)合规性的关键要求,法规明确界定需为每种目标支原体确定阳性检测临界值。验证流程需满足严格的实验设计:每种支原体至少进行三次单独的 10 倍梯度稀释,每次稀释后需制备平行管检测,再确保各稀释浓度获得 24 个检测结果。阳性临界值的判定标准为,该浓度下至少 95% 的试验运行能得到阳性结果,即 24 个样品中需至少出现 23 个有效阳性结果。这一严谨设计旨在确保 NAT 检测方法在实际应用中,能够稳定检出低浓度支原体污染,避免因检测灵敏度不足导致的安全风险,为生物制品质量控制提供可靠保障。
支原体污染来源多样,包括人源、动物源及环境,需多方位监测。安徽免疫细胞产品支原体检测国产替代
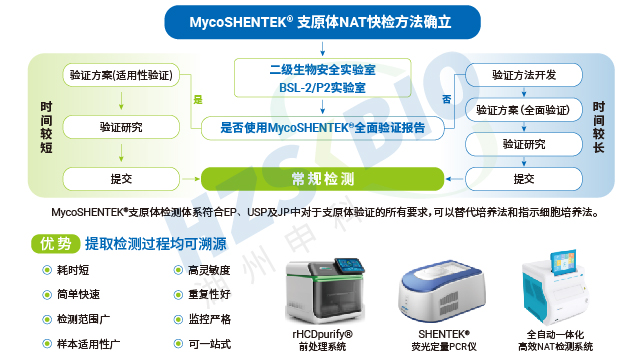
湖州申科构建了具有完备资质的支原体技术服务平台,为企业提供多元化支持。平台拥有 BSL-2/P2 微生物实验室备案资质,遵循 GMP-like 质量体系,具备支原体培养法、指示细胞法与 qPCR 法的检测及验证能力,配备符合药典要求的支原体标准菌株库与高灵敏度培养基(含液体、固体、半流体)。企业已通过 ISO13485:2016 质量管理体系认证(证书号 MD 709873),检测中心获得 CNAS 认证(注册号 CNAS L21942),符合 ISO/IEC 17025:2017 标准,具备国际互认资质。平台可提供多元化技术服务,包括支原体 qPCR 法检测能力建立、实验员能力考核、质量体系与流程搭建、实验室设计方案优化,以及样品检测(三种方法)、样品适用性验证、方法学验证、传统法与 qPCR 法比对、特殊菌株定制生产等,申报阶段可配合客户与监管机构完成现场审计。
湖南重组药物支原体检测试剂盒湖州申科支原体检测验证菌株涵盖 10 余种,满足不同检测场景验证需求。
湖州申科围绕 USP 支原体培养法,提供技术服务,满足生物制剂企业的合规需求。服务内容包括两项:一是支原体培养法样品检测服务,严格遵循 USP 63 标准流程执行,确保检测结果合规有效;二是支原体培养法样品适用性验证服务,针对供试品特性开展定制化验证,保障检测方法与样品的适配性。所有检测与验证完成后,公司将出具标准化报告,报告可直接用于项目申报、审批、工艺优化等场景,为企业产品质量控制提供依据。通过专业的技术服务,帮助企业规避合规风险,确保生物制剂生产全流程的质量与安全性,助力企业高效推进产品研发与上市进程。
支原体 NAT 检测的特异性验证面临主要挑战:需设计覆盖多种支原体的 PCR 引物,而覆盖范围越广,越可能因支原体与革兰氏阳性菌的系统进化相关性,出现交叉检测现象,影响结果准确性。因此,特异性验证需重点排查非目标微生物的干扰,确保检测结果的专一性。稳健性验证同样关键,需证实检测方法在人为引入的微小参数变化(如反应温度、试剂浓度波动等)下,仍能保持结果稳定,避免因实验条件差异导致的检测偏差。这两项验证直接决定了 NAT 方法在不同实验室、不同操作场景下的适用性,是其获得法规认可的重要前提。
USP<77> 要求支原体检测NAT法专属性需经生信分析与实际样品验证,排除近缘菌交叉反应。
AdvSHENTEK外源因子全自动核酸检测分析系统凭借优越性能,为支原体检测提供坚实技术支撑。硬件方面,系统温度运行精度≤0.5℃,温度波动控制在 ±0.5℃以内,荧光强度 CV≤3%,确保检测结果的重复性与准确性;4 通道单独运行且支持同步检测,通道可叠加延展,兼顾检测效率与灵活性。软件方面,系统具备三级权限管理、日志审计追踪功能,完全符合 21CFR Part11 法规要求,支持 LIMS 系统连接、USB 数据导出及打印机直接打印,满足企业合规追溯需求。此外,系统运输与储存便捷,仪器可常规运输,检测试剂盒在 2-8℃环境下即可稳定保存,无需特殊冷链条件,进一步提升了产品的实用性与适配性。
支原体检测取样需按产品体积设计,10-1000mL 样品取总体积 1%,保障结果代表性。湖北疫苗产品支原体检测NAT法
耐用性验证需确认方法参数微小变动时,支原体检测结果不受影响。安徽免疫细胞产品支原体检测国产替代
支原体 NAT 检测的准确性与可靠性受多重因素综合影响,需从人员、方法、设备、试剂耗材、实验室环境五方面严格把控。人员需接受专业培训,熟练掌握 PCR 技术理论与实操流程,积累足够经验并具备责任心,确保操作规范性。方法建立需遵循合理流程并完成充分验证,避免因方法缺陷导致结果偏差。设备方面,PCR 仪与前处理设备的性能、精度、稳定性及定期校准至关重要,直接影响检测数据的重复性。试剂耗材需满足要求:前处理试剂能应对复杂样本基质、耐受常见干扰,检测试剂经性能验证且批间稳定性良好,同时需选用质量合格的耗材。实验室需合理布局,建立完善的管理规范与污染防控措施,为检测提供洁净、可控的环境,避免外部因素干扰。
安徽免疫细胞产品支原体检测国产替代