支原体检测 NAT 方法验证需满足全球药典对检测限、专属性、耐用性、可比性等关键指标的要求,且 EP、USP 新草案提出更严格标准。检测限(LOD)方面,EP 要求针对每种支原体确定临界值,需 3 次单独 10 倍梯度稀释、共 24 个检测数据,95% 检出浓度达标;USP 要求标准菌株浓度≤10 CFU/mL 或 100 GC/mL,≥24 次检测数据支持统计分析;ChP 未明确支原体专属要求,但需满足 “微生物检出下限数量” 设定标准。专属性上,EP 建议测试革兰氏阳性菌交叉污染,USP 要求生信分析与实际样品验证结合,ChP 强调外来成分不干扰试验。耐用性方面,EP 需验证试剂浓度、提取与反应程序变化,USP 涵盖设备、反应体系等参数波动。可比性上,EP 要求 NAT 法与药典法 LOD 及特异性对比,替代培养法需达 10 CFU/mL,替代指示细胞法需达 100 CFU/mL,USP 则视样品基质情况要求可比性研究。
湖州申科提供支原体检测技术培训,助力实验室提升检测能力与合规水平。山东细胞疗法产品支原体检测试剂盒
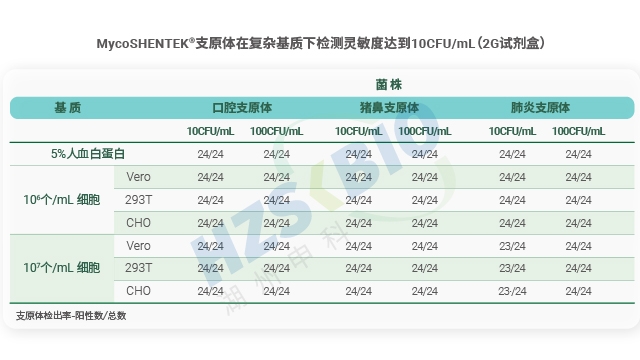
MycoSHENTEK® 支原体 qPCR 检测试剂盒(2G)完全符合 EP 2.6.7 的全部验证要求,其检测灵敏度、特异性、耐用性均按药典标准完成完整性能验证,具备替代培养法和指示细胞培养法的合规资质。该试剂盒针对新型生物制品的检测痛点优化升级,经多种支原体菌株验证,灵敏度稳定达到 10 CFU/mL,满足法规对替代培养法的要求。同时,产品遵循 ISO13485 体系认证和 GMP-like 生产标准,可提供完整的验证报告、质检报告及菌株溯源文件,全程贴合各国药典监管要求,为企业合规检测提供坚实支撑。
福建生物制品支原体检测培养法高浓度质粒样品在进行支原体检测时,需通过浓缩离心预处理,提升支原体检出率。
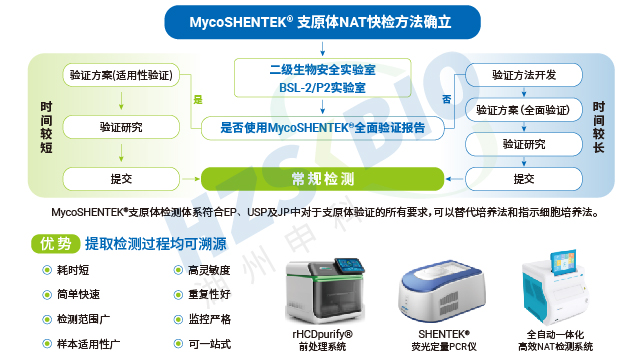
长期以来,支原体检测主要依赖培养法和指示细胞法,且法规通常要求两种方法同时使用,但这两类方法存在明显短板——培养法检测周期长达 28 天,指示细胞法也需较长时间等待结果。随着细胞疗法药物快速发展,其上市周期短、货架期有限的特点,使得传统方法难以满足药物放行的时效要求。核酸扩增技术(NAT)尤其是荧光探针 qPCR 检测方法的出现,凭借检测速度快、特异性强的优势,成为支原体检测的理想替代方案。作为替代方法,NAT 检测需通过严格验证以达到法规要求的灵敏度:检测限达到 10CFU/mL 可替代培养法,达到 100CFU/mL 可替代指示细胞培养法,从而实现快速且可靠的支原体筛查。
细胞和基因治疗领域正加速发展,国内以 CAR-T、间充质干细胞、AAV 基因治疗等新型生物制品势头正盛。这类产品与传统制药差异明显,给支原体检测带来全新挑战:批产量小但批次多,多数待检测样品含高达 10⁷个活细胞,且基质复杂如高蛋白、全血、高浓度质粒等。更关键的是,新型生物制品终末灭菌难度极大,需从起始材料、原物料到全工艺过程严格控污,而支原体污染隐蔽性强、危害大,成为质量安全控制的主要痛点,也推动着检测方法向更高效、抗干扰的方向升级。
支原体污染会改变细胞生理特性,导致生长缓慢、基因表达异常,需严格防控。
为解决支原体检测的污染难题,湖州申科推出AdvSHENTEK® 外源因子全自动核酸检测分析系统 + 一体化支原体检测卡盒的组合方案,以全封闭设计从根源规避污染。该方案只需一步开盖加样,后续流程完全封闭运行,物理隔绝核酸气溶胶污染,搭配 UNG 酶系统可进一步防止环境交叉污染,能节省 100% 污染排查时间。一体化检测卡盒相当于单独的迷你 qPCR 实验室,集成试剂准备、样品制备、扩增、分析全流程,无需复杂分区。同时,方案降低了对实验环境和人员的要求,普通实验室即可开展检测,人员经简单培训就能操作,彻底摆脱了传统 NAT 法对高技能人员和场地的依赖,大幅提升检测结果的稳定性。
EP 新规明确支原体验证菌株 GC/CFU 比值<10,需在指数阶段收获以保障活性与分散性。上海细胞疗法产品支原体检测快速检测
CAR-T 产品支原体检测需在放行前快速完成,湖州申科快速版试剂盒 2.5 小时可出结果。山东细胞疗法产品支原体检测试剂盒
可比性验证是支原体检测 NAT 方法替代传统检测方法的关键依据,法规明确要求需将 NAT 法与药典规定的传统方法进行检测限(LOD)对比。若以 NAT 法替代培养法,需证实其检测限≤10CFU/mL;替代指示细胞培养法时,每种被测支原体的检测限需≤100CFU/mL。对比实验需采用相同样本同步开展 NAT 检测与传统方法检测,通过 CFU 可比性分析,确保两种方法的检测结果具有一致性。这一要求既保障了新方法的检测效能不低于传统标准,又为生物制品企业切换检测方法提供了合规依据,兼顾了效率提升与质量安全。
山东细胞疗法产品支原体检测试剂盒