毕赤酵母(Pichia pastoris)是第二代酵母表达系统中的代表性菌株,是美国FDA认定的GRAS(Generally Recognized As Safe)微生物,具有表达水平高,产物活性好,培养成本低,易扩大为工业化生产等特点。在生物制药领域,酶制剂、胰岛素、表皮生长因子、胶原蛋白等多种生物制剂已经通过毕赤酵母系统进行商业化生产。与其他产品杂质一样,毕赤酵母宿主残留蛋白(HCP)可能对生物制品的安全性和有效性产生不利影响,因此在生产监测、产品放行等过程中需要对其进行定量研究并进行严格控制。SHENTEK®毕赤酵母HCP残留检测试剂盒(一步酶联免疫吸附法)是湖州申科生物自主研发、具有完全自主知识产权的、实现关键试剂全国产化的毕赤酵母HCP通用检测试剂盒。本试剂盒适用于基于GS115、X33等在内的毕赤酵母菌株生产的生物制品中宿主残留蛋白的定量检测,操作步骤少、快速,检测专一性强,性能稳定可靠。
不同 HCP 试剂盒检测结果有差异,企业要评估筛选合适方案。广东工艺特异型宿主细胞蛋白(HCP)残留检测抗体制备
按照美国药典1132章节的要求,HCPs校准品需满足代表性要求,即能覆盖实际工艺产品生产中的HCPs。从HCP免疫检测方法使用目的和预期风险管理要求考虑,满足工艺开发和验证,同时为了应对下游工艺中潜在的异常工艺失效,或工艺变更需求,建议采用上游发酵工艺末端,如澄清处理后工艺点的样本作为HCPs的来源。在实际制备中,可采用空细胞或空载细胞在模拟实际工艺的预定条件进行采集,通过二维电泳或高分辨率质谱等蛋白质组学方法进行模拟工艺和实际工艺下HCPs的代表性表征分析。越靠近下游HCPs蛋白种类越少,也越接近DS中HCPs,但是其可能无法满足工艺开发和验证需求,也无法保证工艺的潜在风险,往往不推荐使用,或只作为上游工艺HCPs免疫检测法的辅助使用。
江苏重组蛋白用宿主细胞蛋白(HCP)残留检测抗体制备为确保HCP ELISA检测产品符合申报要求,湖州申科在试剂盒的全流程开发方案严格按照法规要求。
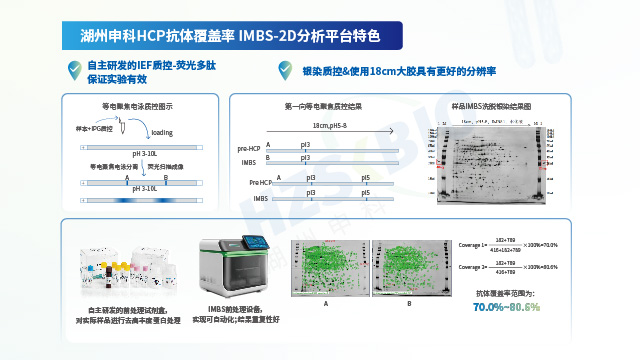
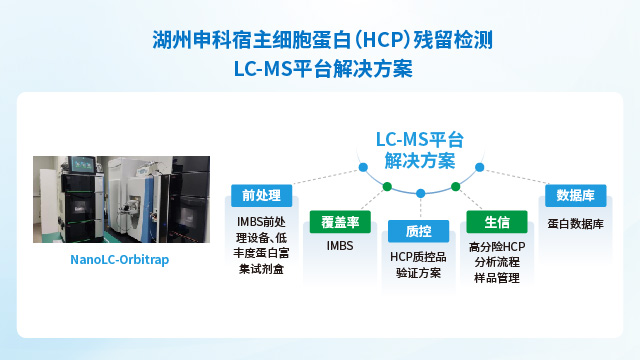
湖州申科生物通过自主可控的供应链体系与严格验证的技术性能,确保HCP检测试剂盒的长期稳定供应与优异的分析能力。一方面,公司实现了关键物料的自研自产:校准品采用冻干工艺大规模制备,可稳定保存10年以上;抗体通过大动物免疫获得,产量可满足≥10,000盒试剂盒的生产需求,保障同批次抗体持续供应超过10年;试剂盒经多批次验证显示良好的批内与批间一致性。另一方面,所有产品参考ICH Q2(R2)和ICH M10法规要求完成验证:以E.coli HCP产品为例,其线性范围(243-1 ng/mL)的R²>0.999,各浓度点回收率偏差≤5%;准确度达81.2%-111.6%,中间精密度CV值5.7%-12.4%;LLOQ低至1.5 ng/mL,且对多种宿主细胞(如CHO、HEK293等)的交叉反应均低于检测限。同时,通过二维电泳(检出826个蛋白点)与质谱法(鉴定2204个蛋白点)双重表征校准品,并采用IMBS-2D(>70%)与IMBS-MS(84.7%)正交技术验证抗体覆盖率,从源头确保检测结果的全面性与可靠性。
各国法规要求必须对生物药品进行分析和纯化,以将宿主细胞蛋白HCP降低到可接受的水平;即使终产品中痕量的宿主细胞蛋白HCP到达患者体内,尚不清楚特定的残留蛋白质杂质是否会影响药物的稳定性或免疫原性。关于HCP的限量标准,美国药典推荐值为终产品的HCP水平1-100 ng/mg;中国药典各论中E.coli菌体HCP应不高于蛋白质总量的0.10% (1000 ng/mg),CHO细胞HCP应不高于蛋白质总量的0.05% (500 ng/mg),假单胞菌HCP应不高于蛋白质总量的0.02% (200 ng/mg)。
部分数据表明,定制化HCP检测试剂盒的检测准确度比商业化试剂盒高,更能满足产品质量控制所需。
对于HCP抗体的纯化方法,目前美国药典1132章节推荐有两种方式,包括protein A或protein G亲和柱层析法和HCP抗原亲和柱层析法。两种方法各有优缺点,均符合监管的要求。在实际使用过程中,这对不同产品可能会导致检测结果的差异。两者方法得到的抗体主要区别是HCP抗体有效含量的占比。HCP抗原亲和柱层析法显然占比高,但是也存在某些HCP抗体丢失的情况,这也会导致针对某些样本的检测结果比前者偏低,需要企业在实际方法建立时进行充分的评估。HCP抗原亲和柱层析法对纯化工艺要求更高,为保证抗体批间一致性,需要重点考察HCP抗原柱制备工艺、柱子的使用寿命、再制备的一致性等问题。
定制化宿主细胞蛋白残留检测试剂盒产生的HCP抗体特异性更好,能检出高风险HCP。上海通用型宿主细胞蛋白(HCP)残留检测抗体覆盖率验证
湖州申科可根据客户要求,快速定制符合用户生产工艺的HCP商业化检测试剂盒,满足用户快速替换的检测需求。广东工艺特异型宿主细胞蛋白(HCP)残留检测抗体制备
为了更好地控制工艺和保证产品质量的稳定,各国监管机构均要求提供使用的宿主细胞蛋白残留检测ELISA试剂盒的抗体覆盖率数据。一般需进行覆盖率分析的场景一般有以下几种情况:①临床II期后,若是继续使用商品化试剂盒,则需要评估试剂盒抗体覆盖率是否可以继续用于质量监控;②临床III期及以后阶段,产品研究者开发了平台化或工艺专属型的HCP监测方法,该类试剂盒在使用前要评估覆盖率水平与商业化覆盖水平的差异;③申报时没有提交覆盖率数据,监管机构可能会对企业提出发补的要求;④产品上市后发生了包括生产场地变更,工艺变更,HCP分析方法变更等因素的变更,研究者则需要评估变更前后抗体覆盖率水平的差异,以及该差异对药品质量与安全带来的影响。
广东工艺特异型宿主细胞蛋白(HCP)残留检测抗体制备