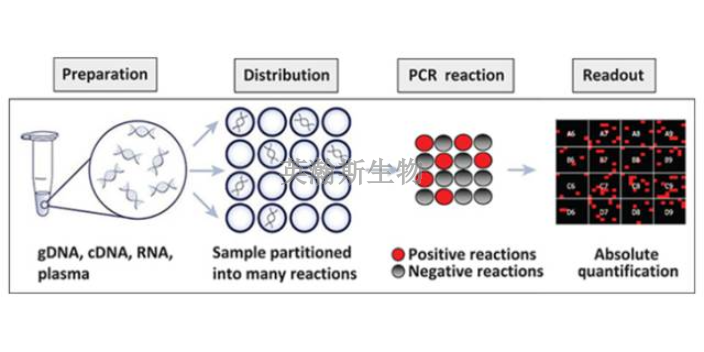
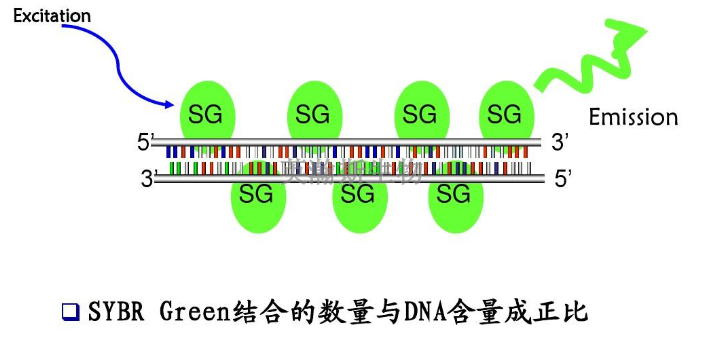
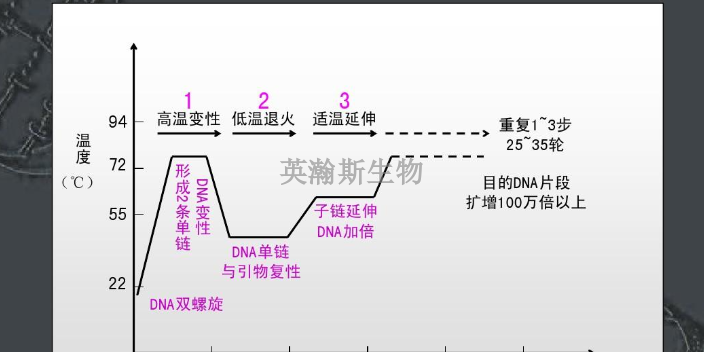
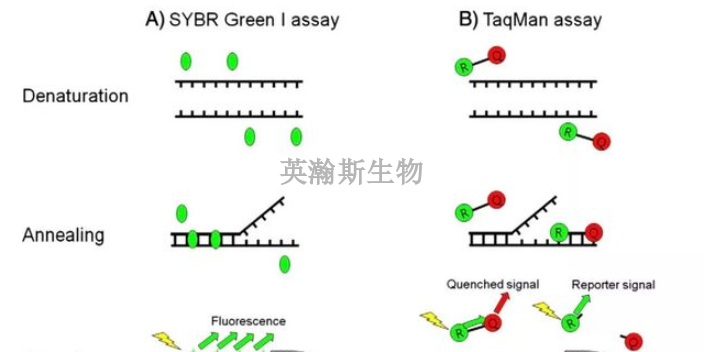
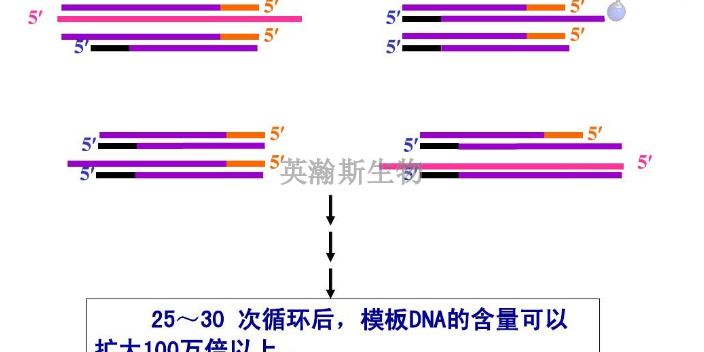
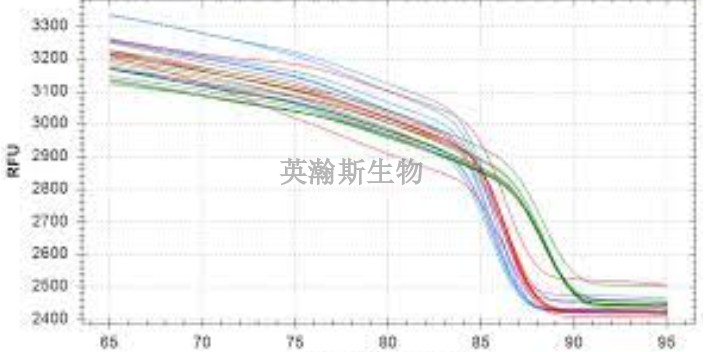
PCR方法主要可以分为三类:终点PCR、qPCR和dPCR。终点PCR终点PCR是较原始、较简单的PCR方法,如今仍在被我们普遍使用。使用者只能在PCR反应结束之后,通过凝胶电泳、毛细管电泳等方法对产物进行检测。终点PCR本身是无法定量的,因为该反应产出的DNA量不一定能反映较初情况。举例来说,不同样品和序列的扩增效率是有差异的。qPCR和dPCR研究者们通过两种途径克服了PCR定量分析的挑战:实时定量PCR(qPCR)和数字PCR(dPCR)。qPCR主要是利用插入性染料或荧光探针(比如TaqMan),人们可以通过监控PCR过程中的荧光强度比较多个样品的DNA水平。数字PCR(dPCR)的基本工作原理很简单,先将样品划分为多个PCR反应,每个反应较多只含有一个模板。然后通过计数正负反应来确定初始样品中模板分子的数量。简述荧光定量pcr的基本原理。贵州细胞pcr
细胞长满80%瓶底或皿底,换培养液,第二天提取RNA(倒掉培养液,5-10ml冰PBS洗涤细胞2次空干)加1ml冰TRizoL,用力振荡培养瓶使细胞完全脱落,加样器吹打(吸入1.5ml离心管,旋涡振荡10秒,室温静置5分钟)加氯仿200ul,漩涡混匀器振荡10秒,冰盒上静置10分钟(40C,8000rpm,5分钟,)吸取上层水相0.5-0.7ml移至另一1.5ml离心管中,加异丙醇0.5-0.7ml(充分混匀,冰盒上静置10分钟,40C,12000rpm,15分)弃上清,加75%冰乙醇1ml,颠倒混匀数次(40C,12000rpm,15分)弃上清,沉淀快速风干。但不要完全干燥,加DEPC处理去离子水50-100ul取5ul作RNA电泳,5ul作RNA定量,余-800C冰箱保存广西常见pcr怎么选择英瀚斯生物,可承接各类荧光定量pcr检测。
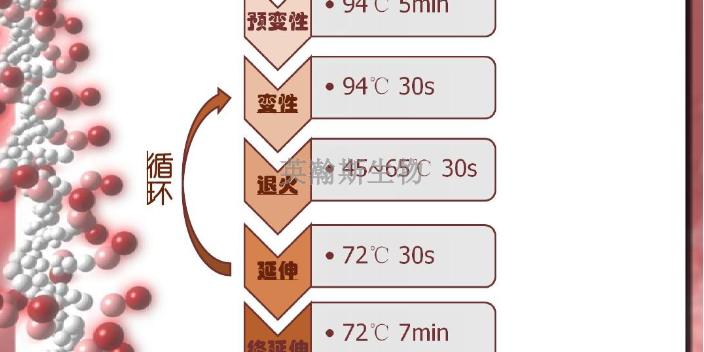
PCR反应的特异性强,其决定因素为:①引物与模板DNA特异正确的结合;②碱基配对原则;③TaqDNA聚合酶合成反应的忠实性;④靶基因的特异性与保守性。其中引物与模板的正确结合是关键。引物与模板的结合及引物链的延伸是遵循碱基配对原则的。聚合酶合成反应的忠实性及TaqDNA聚合酶耐高温性,使反应中模板与引物的结合(复性)可以在较高的温度下进行,结合的特异性明显增加,被扩增的靶基因片段也就能保持很高的正确度。再通过选择特异性和保守性高的靶基因区,其特异性程度就更高。
PCR出现假阳性的原因分析。引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。靶序列太短或引物太短,容易出现假阳性。需重新设计引物。靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。这种假阳性可用以下方法解决:①操作时应小心轻柔,防止将靶序列吸入加样器内或溅出离心管外。②除了酶及其他不耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管均应一次性使用。③必要时,在加标本前,反应管和试剂用紫外线照射,以破坏存在的核酸。二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。pcr检测有哪些操作步骤?
PCR引物设计的原则PCR反应中有两条引物,即5′端引物和3′引物。设计引物时以一条DNA单链为基准(常以信息链为基准),5′端引物与位于待扩增片段5′端上的一小段DNA序列相同;3′端引物与位于待扩增片段3′端的一小段DNA序列互补。(1)引物设计的基本原则引物长度:15-30bp,常用为20bp左右。引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。ATGC比较好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列参照。引物内部不应出现互补序列。两个引物之间不应存在互补序列,尤其是避免3′端的互补重叠。引物与非特异扩增区的序列的同源性不要超过70%,引物3′末端连续8个碱基在待扩增区以外不能有完全互补序列,否则易导致非特异性扩增。引物3‘端的碱基,特别是较末及倒数第二个碱基,应严格要求配对,比较好选择是G和C。引物的5′端可以修饰。如附加限制酶位点,引入突变位点,用生物素、荧光物质、地高辛标记,加入其它短序列,包括起始密码子、终止密码子等。荧光定量pcr和实时定量pcr的区别?浙江有哪些pcr扩增
细胞上清提取RNA进行PCR检测。贵州细胞pcr
PCR实验技术中的反向PCR(InversePCR)介绍:反向PCR起初设计用于确定临近未知区域的序列。反向PCR有助于研究一个基因的启动子序列;致*染色体重排如基因融合、异位或转移;及病毒基因的整合。之所以称为反向PCR,是因为引物设计用于向两边延伸而不像常规PCR中朝着彼此延伸。如今,反向PCR常用于定点突变,复制一个具有预期突变的目的质粒。在研究基因组DNA未知序列的传统实验流程中,限制性内切酶消化和连接先于反向PCR,接着对PCR扩增子进行测序。对于gDNA消化,需选择一种限制性内切酶进行酶切以获得合适长度可自连的片段。同时,所选择的内切酶不应该切割已知序列,所以连接发生于侧翼的未知序列之间。使用低浓度的酶切DNA的片段以助于片段自连而非多个片段连接。片段自连完成后,通过反向PCR扩增DNA的位置区域。获得的扩增子两端包含一部分已知序列。之后通过测序以鉴定临近已知序列的未知区域。贵州细胞pcr
南京英瀚斯生物科技有限公司属于医药健康的高新企业,技术力量雄厚。公司致力于为客户提供安全、质量有保证的良好产品及服务,是一家有限责任公司(自然)企业。公司拥有专业的技术团队,具有实验外包,动物模型构建,细胞分子实验,病理检测等多项业务。英瀚斯生物自成立以来,一直坚持走正规化、专业化路线,得到了广大客户及社会各界的普遍认可与大力支持。